The structure of myoglobin; the ribbon represents the path of the polypeptide chain and is color coded blue at the N-terminus, running through to red at the C-terminus. Note the organization into many helical segments.
| The polypeptide chain forms a backbone structure in proteins: | 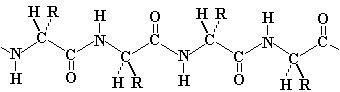 |
| On first inspection, this structure appears to be connected entirely
by single C-C or C-N bonds. It should therefore be as flexible as a
simple hydrocarbon chain.
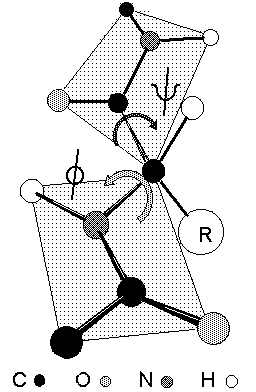
Note that flexing in a covalent structure does not occur by bending bonds, and the normal tetrahedral or trigonal planar bond angles are maintained. Instead, different shapes are obtained by torsional rotation about the axis of the bonds: |
 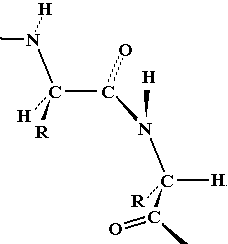 |
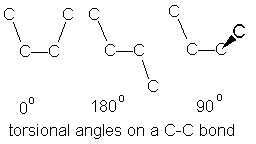 |
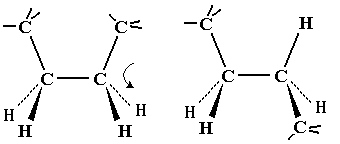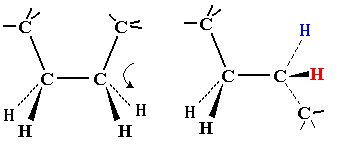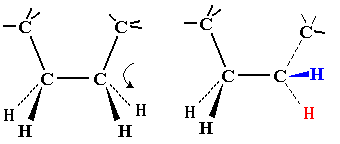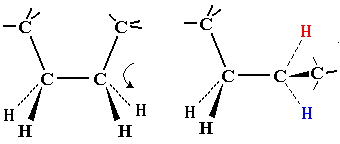 |
All polypeptides can adopt a form which is flexible, but random and disordered in bond orientation. This is called the denatured state of the protein. Proteins become denatured at elevated temperature or in the presence of disruptive solvents. Because there is no orderly arrangement, denatured protein is non-functional. Most proteins also have an ordered arrangement called the native state. Biochemists often work with proteins in ice baths to keep the temperature down and preserve the native state.
alpha-keratin - protein of hair, skin and wool;
beta-keratin or fibroin - spider and silkmoth silk.
The keratins were studied by X-ray fiber diffraction, a technique in which X-rays are reflected off a regular repetitive macromolecule array in the protein fiber, forming a characteristic pattern if the repeat spacing of the sample is comparable to X-ray wavelengths. If the X-ray wavelength is known, the size of the repeat pattern in the protein can be calculated.
For alpha-keratin, periodic repeat distances were 1.5 and 5.4 angstroms. The diffraction patterns also showed characteristic missing reflections that suggested a helical structure.
For fibroin or beta keratin, periodic repeats were 3.5 and 7.0 angstroms.
The structures having these repeat distances were solved by Linus Pauling. Pauling understood the importance of knowing exact atomic radii, bond lengths and angles and used these to create exact scale models of possible structures.
However, the simple single-bonded peptide chain was so flexible that it was not initially apparent why specific structures would form.
| By measuring bond lengths, Pauling first deduced that the peptide bond had double bond character, due to the second resonance form of the amide. | 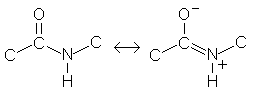 | ||||||||||||
The shape of the peptide chain can be defined by the three consecutive bond torsional angles
Peptide bonds are almost invariably fixed at omega =
180o or trans based on the relative alignment of C |
C
|
The rigidity of the peptide bond limits the number of arrangements that Pauling's models could fit without distorting bonds or forcing atoms closer than van der Waals radii would allow. Without this constraint, the peptide would be free to adopt so many structures that no single consistent pattern would emerge. By reducing the degrees of freedom, a well defined set of states emerges.
Pauling found that two general patterns conformed with atomic geometry:
| 1) an extended state for which angles phi = -135o and psi = +135o; the polypeptide chain alternates in direction, resulting in a zig-zag structure for the peptide chain. Note the shaded circle around R; the extended strand arrangement also allows the maximum space and freedom of movement for a side chain. The repeat between identically oriented R-groups is 7.0 Å, with 3.5 Å per amino acid, matching the fiber diffraction data for beta-keratins. | 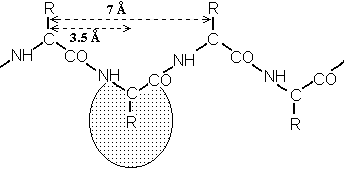 |
|
2) a helical state in which the phi and psi were roughly -60O, twisting repeatedly in the same direction. The helical form models could be built with varying degrees of twist, but one model fit the atomic dimensions especially well: Alpha helix: has 3.6 amino acids per turn of the helix, which places the C=O group of amino acid #1 exactly in line with the H-N group of amino acid #5 (and C=O #2 with H-N #6). |
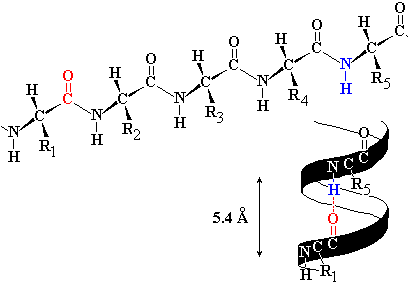 |
| The alignment and spacing is ideal for a hydrogen bonds to form
C=O:---H-N.
The figure shows an alpha helix with side chains omitted for clarity. C = gray, N= blue, O = red. Hydrogen bonds are shown as dotted lines in the figure, and hydrogen bonding would make this structure especially stable. The distance separating each turn of the helix was 5.4 Å, matching periodic repeats in alpha keratin, hence the name alpha helix. Since 5.4 Å / 3.6 is 1.5 Å, the alpha-helix has an amino acid every 1.5 Å, matching the minor periodic repeat of alpha keratin. The alpha helix was also found to be exclusively right handed; a left handed arrangement (phi and psi = +60o) has similar dimensions, but positions the amino acid side chains next to the C=O group, making the structure over-crowded. The right handed version of the helix places side chains next to the much smaller N-H, for a better fit. |
 |
|
Myoglobin was the first globular protein whose structure was worked out from X-ray diffraction by protein crystals. The periodic repeats characteristic of alpha helix were recognised, and this helped determine the structure shown, in which 70% of the polypeptide is alpha-helical. |
|
Other helices discovered by Pauling include the 2.27 helix, the
310 helix and the pi helix.
310 refers to a helix of 3
amino acids per turn, with hydrogen bonds from #1C=O to H-N #4; the hydrogen
bond closes a loop of 10 atoms.
2.27 is a tight helix of 2.2 amino
acids per turn, a 7 atom loop being closed by the H-bond #1 C=O to H-N #3. The
alpha helix would be 3.613 in this nomenclature; the pi helix is
4.416. Although all these helices could be modelled, experimental
data was only consistent with alpha helix.
In a search of about 30000 helix segments in the protein data base:
In the alpha helix, the C=O---H-N bonds are almost parallel with the helix axis. The basis of hydrogen bonding is strong dipole-dipole interactions, and thus the H-bond dipoles reinforce in the helix, positive end towards the start or N-terminal end of the dipole. H-bonds are skewed relative to the helix axis in the other helices, so the reinforcement of dipoles is less effective.
|
Pauling's extended state model matched the spacing of fibroin
exactly (3.5 and 7.0 Å). In the extended state, H-bonding NH and CO groups
point out at 90o to the strand. |
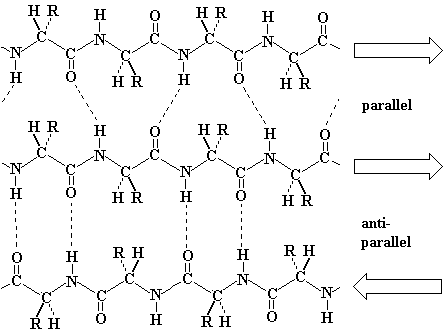 |
Antiparallel beta-sheet is significantly more stable due to the well aligned H-bonds.
| The Pauling model building studies of the early 1950's were followed up by an exhaustive computer search of phi and psi space, carried out in the late 1960's by Ramachandran. The analysis was based on potential energy calculations, i.e. through electrostatic and van der Waals interactions. Ramachandran showed that serious steric interferences occurred between C=O groups and amino acid side chain centred at phi = 120o, and serious interferences between peptide backbone CO and NH occurred at phi = psi = 0o. | 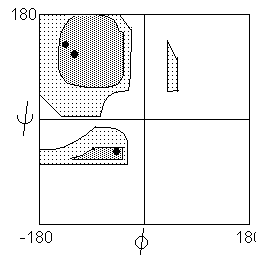 |
There are two regions where interactions may be described as favourable: The region of highest stability is a broad plateau centered at phi = -120O and psi = +135o. This region includes the parallel and antiparallel beta sheet.
The breadth of this region indicates that a degree of variability is allowed to the beta structure. As observed in actual proteins, beta sheets twist and flex, and beta sheets rarely actually conform exactly to the canonical extended strand. Some specialized structures such as polyglycine and polyproline are observed the edge of this zone, where phi = -60o.
A slightly lower but still energetically favoured region is represented by a strip running from phi = -60o to -120o and psi = -60o. The alpha helical conformation lies in this region, while 310 and pi helices flank the favoured region.
In addition to the two favoured regions, two regions were calculated as allowed although not favoured. These include the mirror image of the alpha-helix strip (left handed alpha helix and the isthmus connecting the two favoured regions). The mirror image of the strongly favoured beta sheet plateau turns out to be strongly disfavoured, except by polyglycine (which has a side chain consisting only of a single H atom).
Statistical analysis of observed phi and psi angles in proteins show that most instances lie in or close to the two favoured regions (distribution shown below left), with the exception of glycine, which may adopt conformations corresponding to the mirror image of the favoured states (distribution below right).
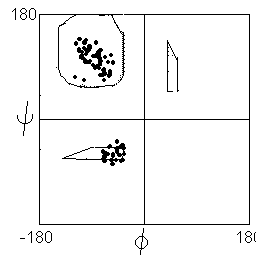 |
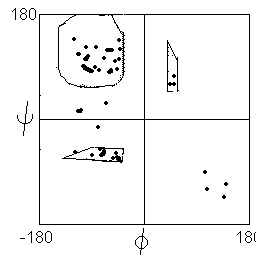 |
Folded proteins contain a considerable proportion of alpha helix or beta
sheet: myoglobin, an alpha-helical bundle, is 70% alpha helix;
other
proteins may contain beta strands or a mix of alpha and beta. An alpha helix has
all its amino acids repeating the same phi/psi conformation. Likewise for beta
sheet, with a bit more variability due to the broad plateau in the Ramachandran
plot.
The remainder of the polypeptide exists either in the form of tight turns or loops, connecting elements that link one segment of secondary structure to the next. Tight turns consist of 4-5 amino acids in a fairly well defined structure - it's just not regular and repetitive like alpha helix or beta sheet. Loops (or random coil) are completely non-repetitive.
Alpha helix may be considered the default state for secondary structure. Although the potential energy is not as low as for beta sheet, H-bond formation is intra-strand, so there is an entropic advantage over beta sheet, where H-bonds must form from strand to strand, with strand segments that may be quite distant in the polypeptide sequence.
The main criterion for alpha helix preference is that the amino acid side chain should cover and protect the backbone H-bonds in the core of the helix. Most amino acids do this with some key exceptions:
| alpha-helix preference: | Ala,Leu,Met,Phe,Glu,Gln,His,Lys,Arg |
The extended structure leaves the maximum space free for the amino acid side chains: as a result, those amino acids with large bulky side chains prefer to form beta sheet structures:
| just plain large: | Tyr, Trp, (Phe, Met) |
| bulky and awkward due to branched beta carbon: | Ile, Val, Thr |
| large S atom on beta carbon: | Cys |
The remaining amino acids have side chains which disrupt secondary structure, and are known as secondary structure breakers:
| side chain H is too small to protect backbone H-bond: | Gly |
| side chain linked to alpha N, has no N-H to H-bond; rigid structure due to ring restricts to phi = -60o; |
Pro |
| H-bonding side chains compete directly with backbone H-bonds | Asp, Asn, Ser |
Clusters of breakers give rise to regions known as loops or turns which mark the boundaries of regular secondary structure, and serve to link up secondary structure segments.
There are various schemes that give the amino acids numerical weights or rankings for their preferences, and several computer programs can predict the secondary structure from the given sequence. The simplest such scheme of Chou and Fasman, Ann. Rev Biochem. 47 258 (1978), examined the statistical distribution of amino acids in alpha helix, beta sheet and turns or loops, using a set of known protein structures from the protein databank.
A novel sequence can then be scanned, and the tendency of each portion of the sequence to form secondary structure is assessed. An important factor is that the final secondary structure is the consensus of all amino acids in a region. A minimum of 4 amino acids out of 6 should show alpha preference, or 3 out of 5 beta preference, or clusters of 2-3 breakers in a sequence of 4 are needed to set the secondary structure in any region, and individual misfits adopt the secondary structure of their neighbours. If you have one or two helix preference amino acids scattered in a majority of beta sheet former, the structure adopted is entirely beta sheet.
More recent prediction schemes take advantage of larger data sets to examine amino acid preference for different regions in a helix or different positions in a tight turn. In addition, sequences of homologous proteins may be compared. The rationale is that highly conserved amino acids contribute more to the three dimensional structure than unconserved, and different weightings can be introduced to the statistical analysis.
The accuracy of prediction has risen from about 55% using the simple Chou-Fasman method, where the tendency is to overpredict, to about 80% using current methods. Current methods are now so complex that they are not attempted by individuals. Instead, one submits a sequence as an e-mail to certain web sites, and the results are returned in a matter of a few hours.
http://www.bmm.icnet.uk/people/rob/CCP11BBS/
C.Geourjon & G. Deleage, Protein Engineering, 7, 157-164 (1994).
The accuracy level of 80% usually represents good prediction of the occurence of secondary structure elements, with errors in the exact position of boundaries. It may not be possible to improve much further, since it is believed that secondary structure adjusts itself due to interactions that arise in the folded form of the protein, which a sequence scan approach can't account for.
Another useful link: the Principles of Protein Structure web course at Birkbeck College, UK. This is a very extensive web site, essentially representing a semester graduate course in protein structure.
http://PPS97.cryst.bbk.ac.uk/index.html
A quick illustrated review for exploring in your own time: